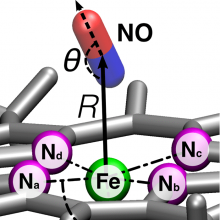
Multidimensional potential energy surfaces based on reproducing kernel-interpolation are employed to explore the energetics and dynamics of free and bound nitric oxide in myoglobin (Mb). Combining a force field description for the majority of degrees of freedom and the higher-accuracy representation for the NO ligand and the Fe out-of-plane motion allows for a simulation approach akin to a mixed quantum mechanics/molecular mechanics treatment. However, the kernel-representation can be evaluated at conventional force-field speed. With the explicit inclusion of the Fe-out-of-plane (Fe-oop) coordinate, the dynamics and structural equilibrium after photodissociation of the ligand are correctly described compared to experiment. Experimentally, the Fe-oop coordinate plays an important role for the ligand dynamics. This is also found here where the isomerization dynamics between the Fe–ON and Fe–NO state is significantly affected whether or not this co-ordinate is explicitly included. Although the Fe–ON conformation is metastable when considering only the bound 2 A state, it may disappear once the 4 A state is included. This explains the absence of the Fe–ON state in previous experimental investigations of MbNO.