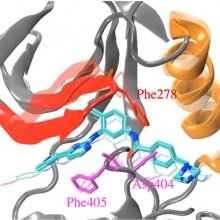
Gleevec is a potent inhibitor of Abl tyrosine kinase but not of the highly homologous c-Src kinase. Because the ligand binds to an inactive form of the protein in which an Asp-Phe-Gly structural motif along the activation loop adopts a so-called DFG-out conformation, it was suggested that binding specificity was controlled by a “conformational selection” mechanism. In this context, the binding affinity displayed by the kinase inhibitor G6G poses an intriguing challenge. Although it possesses a chemical core very similar to that of Gleevec, G6G is a potent inhibitor of both Abl and c-Src kinases. Both inhibitors bind to the DFG-out conformation of the kinases, which seems to be in contradiction with the conformational selection mechanism. To address this issue and display the hidden thermodynamic contributions affecting the binding selectivity, molecular dynamics free energy simulations with explicit solvent molecules were carried out. Relative to Gleevec, G6G forms highly favorable van der Waals dispersive interactions upon binding to the kinases via its triazine functional group, which is considerably larger than the corresponding pyridine moiety in Gleevec. Upon binding of G6G to c-Src, these interactions offset the unfavorable free energy cost of the DFG-out conformation. When binding to Abl, however, G6G experiences an unfavorable free energy penalty due to steric clashes with the phosphate-binding loop, yielding an overall binding affinity that is similar to that of Gleevec. Such steric clashes are absent when G6G binds to c-Src, due to the extended conformation of the phosphate-binding loop.